

Röntgendiffrakciós módszerrel a fehérje minden egyes atomjának a helye meghatározható. Ez alapján grafikus programmal a fehérjét könnyűszerrel ábrázolhatjuk és megvizsgálhatjuk, hogy mely aminosavak léphetnek kölcsönhatásba, hol vannak nagyobb üregek vagy vannak-e a fehérjében kismolekulák megkötésére képes részletek. A röntgendiffrakciós méréshez azonban kristályosítani kell a fehérjét, ami nem egyszerű feladat, hiszen egy-egy új fehérjetípus esetén mindig meg kell találni az ideális körülményeket ahhoz, hogy a kristály szabályosan növekedjék. Membránba ágyazott fehérjéknél különösen körülményes lépés a kristálynövesztés, hiszen a fehérjéről leoldjuk természetes közegét, a sejtmembránt, így a fehérje nehezen veszi fel újra a szerkezetet.
Ha nem szeretnénk fehérjekristállyal dolgozni, akkor lehetőség van az NMR módszerrel történő szerkezetmeghatározásra is. Ehhez elég az oldott, tiszta fehérje, sőt a mérés eredményeként nem is „állóképet” kapunk, hanem mindjárt több felvételt. A röntgendiffrakciós módszerrel szemben, - amely a fehérjének azt az alacsony energiájú állapotát tükrözi, amelyben kristályosodott -, az NMR felvételből a mozgó fehérjeláncok számos lehetséges állapota, illetve azok átlaga is visszafejthető. Az NMR eljárást viszont egy bizonyos mérethatáron túl már nem lehet alkalmazni, márpedig a receptorok transzport fehérjék és egyéb, a jelátvitelben fontos egységek már ebbe a tartományba esnek.
Hogyan juthatunk akkor közelebb a fehérjeszerkezethez, pusztán a fehérjealkotó aminosavak sorrendje alapján?
A fehérjeszerkezetek központi adatbázisában (Protein Data Bank, PDB) ma már több, mint 46 000 fehérjeszerkezetet található. Ezek aminosavsorrendjét elemezve számos becslő módszert fejlesztettek ki, amelyekkel egy-egy ismeretlen fehérjéről megjósolható, hogy hol találhatók benne helikális, vagy redőzött szakaszok, membránt átszelő láncok, hol várható rajtuk foszforillálódás vagy egyéb módosítás. Az utóbbi időben egyre több fehérjéről derül ki, hogy ú.n. rendezetlen szakaszokat tartalmaz, amelyek nem is rendelkeznek időben állandó szerkezettel, ezek becslésére is lehetőség van. A fenti információk alapján már hozzávetőleges képet kaphatunk az áhított fehérjéről, de ez általában még mindig nem elég. A rendezett szakaszokat tartalmazó fehérjékről ismert, hogy az aminosav sorrendjük egyértelműen meghatározza a térszerkezetet. Vagyis, ha ismerjük a szekvenciát, az csak egyféle szerkezetet képes felvenni. Ennek alapján, hasonló aminosav sorrendű fehérjéknek általában hasonló a szerkezete, éppen ezen a tényen alapszik fehérjemodellezés egyik módja a homológiamodellezés.
Az eljárás során olyan fehérjét választunk mintának, amelynek a szekvenciája hasonló a vizsgált fehérjéhez, de a szerkezete is ismert. Ez lesz a mintadarab a modellezés során. Minél jobban hasonlít az ismert és a modellezendő fehérje aminosav sorrendje, annál jobban bízhatunk a modellünkben. Fontos még, hogy megtaláljuk a két szekvencia között a lehető legjobb megfeleltetést: azt az illeszkedést, ahol minél több azonos aminosav illeszkedik egymáshoz. Azokban az esetekben, ahol az aminosavak eltérőek, igyekszünk a „hasonlókat” (nagyméretűt a nagyméretűhöz, polárost a polároshoz stb). illeszteni.

Példa két hasonló enzim egy-egy részletének illesztésére. A betűk az aminosavak egybetűs kódját jelölik, sárgával és az illesztés alatt csillaggal az azonos aminosavakat, ponttal a hasonló típusú aminosavakat jelöltük.
A két fehérjelánc hossza többnyire eltérő, ilyenkor lesznek olyan szakaszok, amelyeket egyáltalán nem tudunk megfeleltetni egy másiknak, így az illesztésbe az egyik szekvencián üres helyeket kell betoldanunk. A lehető legpontosabb illesztés a modell egyik kulcsa, amit ma már automatizált számítógépes programok segítségével végzünk. Egy-egy manuális korrekció azonban sokat javíthat az illesztésen. Ha kísérletek alapján feltételezhető, hogy két aminosav kölcsönhatásban van, vagy egymáshoz közel helyezkedik el, akkor korrigálhatjuk az illesztést úgy, hogy az ismeretlen szerkezetű fehérje szekvenciáját az ismert fehérje vázára húzva már teljesüljön a kísérletben talált feltétel. Ha megalkottuk a modell vázát, akkor számítógépes eljárással elrendezhetők az oldalláncok, majd ellenőriznünk kell, hogy „élhető”-e a szerkezet: nincsenek-e ütközések, vagy kirívó, gyanús részletek a molekulán.
A fenti illesztés eredményeként a következő szerkezeti részletet kapjuk.
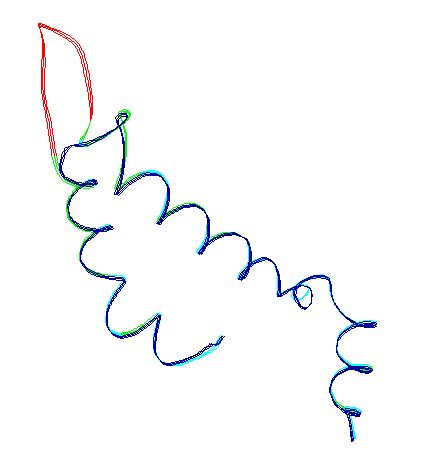
A zöld színnel jelölt szakaszokon a modellezendő fehérje aminosavai nagy bizonyossággal ráhúzhatók az ismert szerkezetű fehérje vázára. A pirossal jelölt betoldott szakasz a modell legbizonytalanabb része, ezt finomítanunk kell.
Ha mindent rendben találunk, a fehérjemodellen szemügyre vehetjük a kialakult csatornákat, lehetséges kötőhelyeket, a működés közben elmozdulni képes szakaszokat és olyan kísérleti eredményekre találhatunk szerkezeti magyarázatot, amelyekre pusztán az aminosav sorrend alapján esetleg sosem gondoltunk volna. Azt a kérdést azonban, hogy sikeres, élethű lett-e a modellünk, véglegesen a kísérletek és a szerkezetvizsgáló laboratóriumokban meghatározott eredmények döntik el.




Utolsó kommentek